 收藏
收藏 询价
询价数据分析
快速帮您解决工作中遇到的问题

常见问题
数据分析
1. 异常图像分析
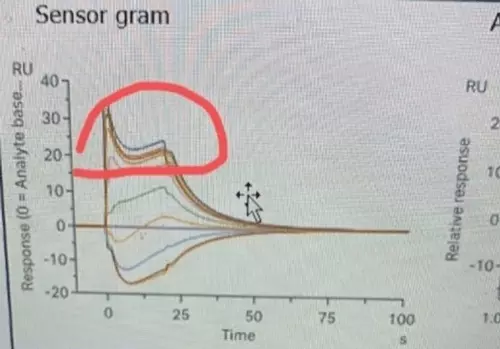
传感图倒置是什么原因,如何处理?
出现这种情况的影响因素分不同类型,需要逐一排查,首先可以确认必然是参比的信号高了导致的。什么导致它高了?
- 最基础的,设置程序或偶联时是否选错通道,尤其捕获法时。
- 看流动相样品是否有高折光物质比如甘油、蔗糖、咪唑(置换buffer)。
- 看看流动相样品本身存在缓冲体系是不是跟实验用buffer差异大(置换buffer)。
- 看看是不是流动相样品粘稠或者浓度太高。
- 如果是涉及到有机溶剂那要看有没有做溶剂校正。
- 非特异结合(提高盐离子浓度,换buffer,分析物配体对调后实验)。
请问,在跑小分子,浓度都提高几百微摩尔级别,但是拟合曲线还是呈线性,算不出KD值,这是怎么回事?是否有非特异性结合?
想查看是否有非特异性结合,请点击分析软件中左侧一列的Bingding to reference,看看是否出现明显的随浓度而增加的信号上升。
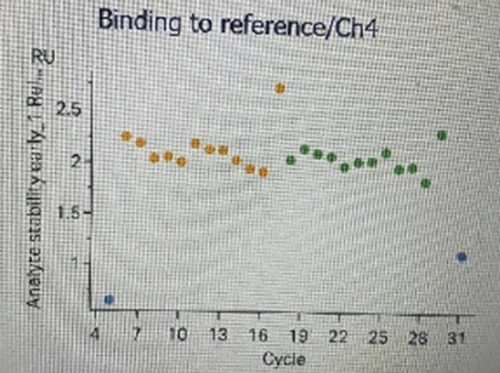
分析结果显示,Binding to reference出现有规律的上升,这是什么现象?什么原因造成的。
想查看是否有非特异性结合,请点击分析软件中左侧一列的Bingding to reference,看看是否出现明显的随浓度而增加的信号上升。
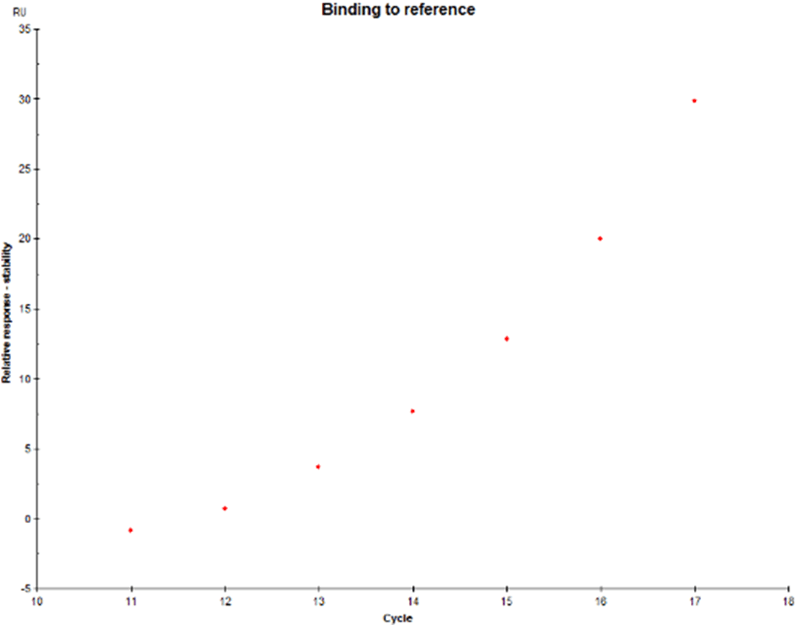
- 如图所示,很明显,随着浓度的升高,Binding to reference的数值也随之上升,这是非特异结合的表现。
- 造成的原因可能是,分析物与通道结合,蛋白聚集等。
参比通道非特异结合强,如何处理?
- 一般建议对活化后再封闭参比通道
- 其他降非特异结合的方式,尝试调整buffer条件,pH,盐浓度,去垢剂浓度等
- 使用NSB reducer
- 偶联伪装配体(例如BSA)
在分析数据时,发现有曲线大幅度波动,这是什么原因造成的?
想查看是否有非特异性结合,请点击分析软件中左侧一列的Bingding to reference,看看是否出现明显的随浓度而增加的信号上升。
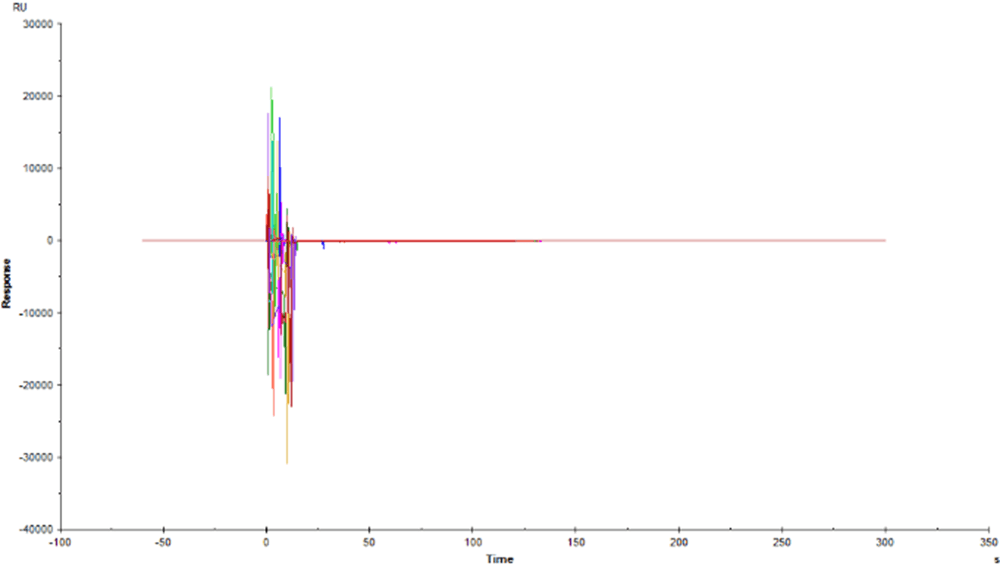
- 从图中,我们可以看出,曲线有上万RU的起伏,最大可能是气泡造成的效应
- 样品进样前,可以把样品离心一下,Buffer要脱气
分析结果中的图像出现向上漂移现象,原因是什么?
分析结果中的图像出现向上漂移现象,原因是什么
关注零浓度情况(Biacore Insight软件关注Blank情况),该图像往往是由于零浓度信号不正常造成的。
- 小分子实验:高偶联情况下(CM5>10000RU),在偶联完成后可以放置一段时间平稳后再进行实验
- 捕获实验:标签结合情况一般的情况下,会导致配体亲和效果不佳;可选择更优的捕获分子或使用商业化捕获试剂盒,进行配体捕获。
- 高亲和实验:高亲和力、慢解离实验中,不适合使用捕获方法,推荐使用直接偶联方法进行实验。
- 没有其他问题情况下,建议在设置浓度梯度时,多设置几个零,最终分析结果时,挑选无误的那个零做扣除。
2. 小分子-蛋白互作
直接偶联法种提高蛋白偶联量的方式?
- 正式偶联前进行pH scouting的实验
- 确认蛋白等电点是否为酸性蛋白
- 确认蛋白纯度后,可提高配体蛋白进样浓度,延长配体蛋白进样时间
- 确认该蛋白是否氨基过少
- 是否活化不充分
- 活化峰在4500-5500RU左右
- 火花前后的信号差异200RU左右
- 芯片改用CM7
预富集效果良好,但实际偶联量极低,什么原因,如何改善?
- 可能活化剂失活
- 可能蛋白的精氨酸含量低,转化率低(若是该原因解决办法:可以改成捕获法)
其中5个浓度都正常,但最高浓度突然响应值很高,为什么?

小分子样品高浓度下,样品会有聚集,产生异常高信号
偶联了一个二聚体蛋白到CM7芯片,偶联量17000RU,基线一直不平而且上升是什么原因呢?
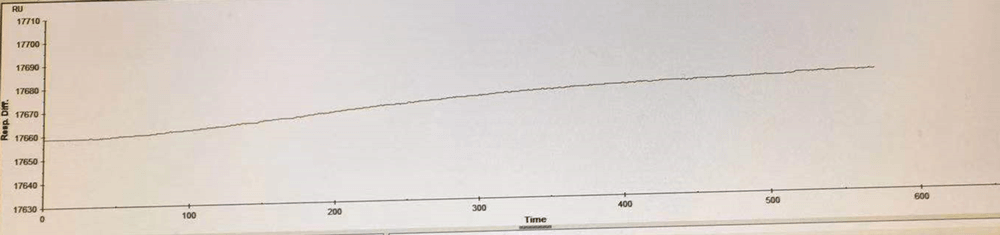
有聚集。偶联浓度不要太高,Running buffer中要有EDTA
用NTA chip研究药物和蛋白的反应 ,药物溶液里有DMSO,做了多次,图形里面一直有这个Peak,怎么才能去掉Peak?
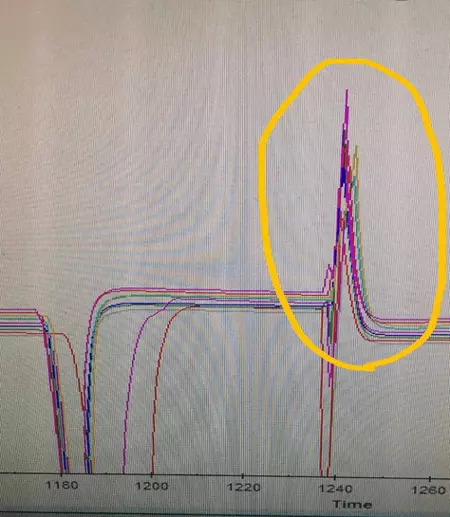
该Peak是由于DMSO等有机溶剂的折光导致,药物溶液中有DMSO时,样品和buffer要采用相同浓度的dmso,并对其进行溶剂校正,得到真实结果。
溶液切换时,折光差异变化较大,如图(小分子;CM7;PBS buffer),为什么?
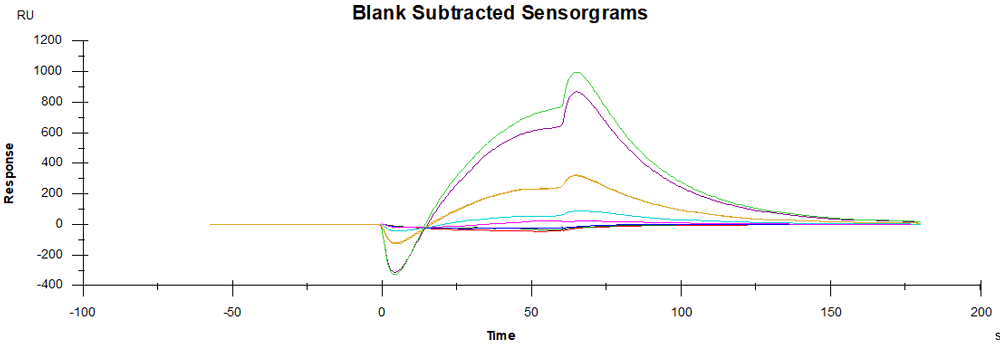
溶液切换时,折光差异变化较大,如图(小分子;CM7;PBS buffer)
- 原因:最高浓度的样品稀释的倍数小了,导致buffer 和样品的折光差异大,信号有差异
- 调整:最高浓度的样品增加稀释的倍数;提高流速;避免跨通道扣减
Biacore跑出来的曲线出现了不规则的形状,无论在结合段还是解离段,什么原因造成的,如何处理?
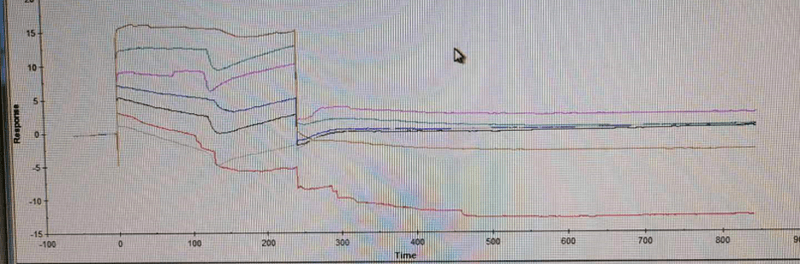
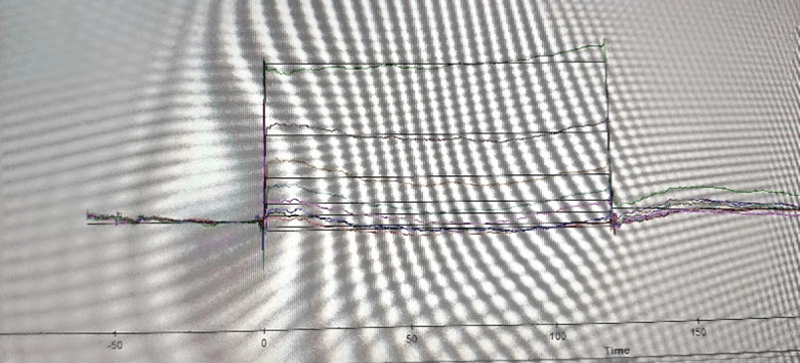
建议先查看分析物溶解度,优化分析物溶解度,然后再跑一次,看看是否还是出现这种现象。
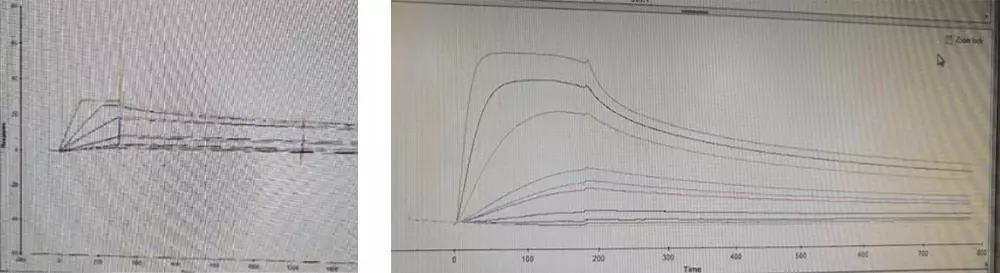
如图所示,出现不规则图像如何调整?
1. 先看是否存在非特异性,binding to reference如下图平稳则无。
2. 再看buffer是不是有比较特殊的成分存在。
3. 蛋白-蛋白互作
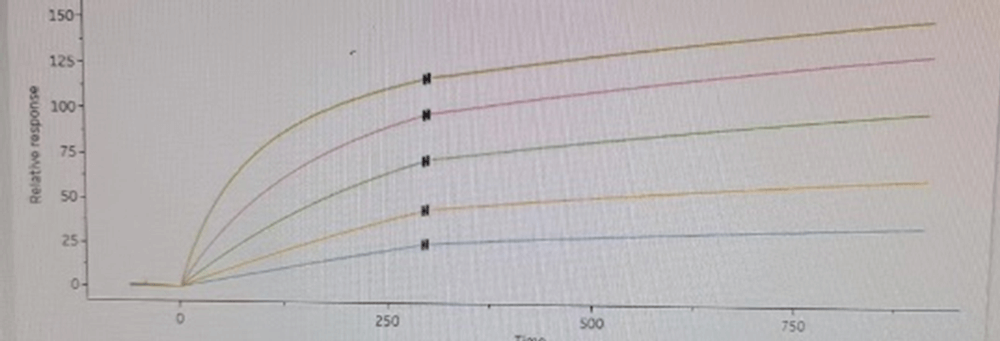
物质迁移的定义是什么?如何避免?
- 物质迁移效应是指分析物分子受到扩散速率的限制,从溶液本体向芯片表面供应不足,造成分析物浓度递减的现象。
- 如图所示,很明显在结合段,特别是高浓度结合段,结合曲线都是直接以直线形式上升的,并没有出现典型的高浓度弯曲,这就是典型的物质迁移效应。
- 解决的方法有:
- 降低偶联量(做大分子互做时,Rmax要控制在50RU以内;在做小分子互做时,Rmax要控制在100RU以内)
- 提高分析物的流速(大于等于30ul/min)。
抗原抗体动力学检测,拟合效果不好,但是所有的QC都很好,请问如何优化调整?
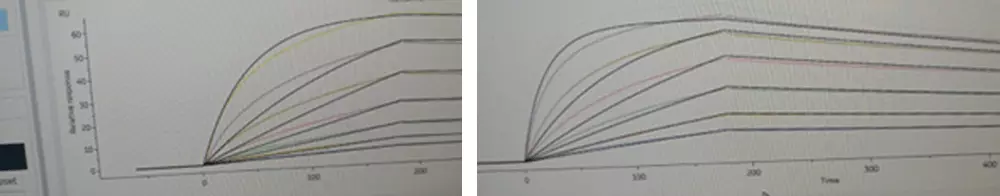
- 该结果已经足够说明互作,并且结果是可靠的。
- 如果需要进一步提升拟合结果的质量,建议如下:可将RI 改成Fit local进行拟合即可。
4. 捕获法
捕获之后响应值一直在掉,该怎么处理?
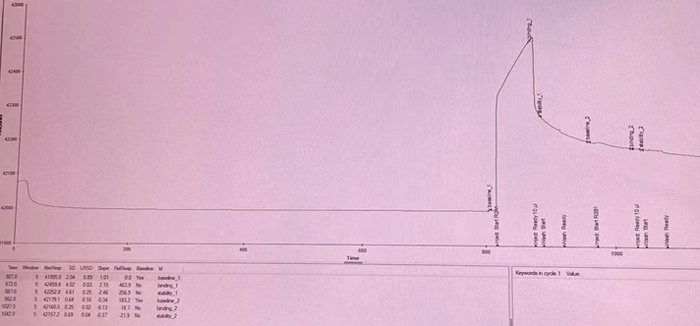
- wait基线变平,等capture level稳定后再进sample。
- 降低捕获量。
- 进行高浓度、短时间的捕获。
使用Biacore 8K时,捕获抗体走抗原,发现曲线没有明显饱和,而且分析的结果也超出仪器范围之外,请问怎么调整?
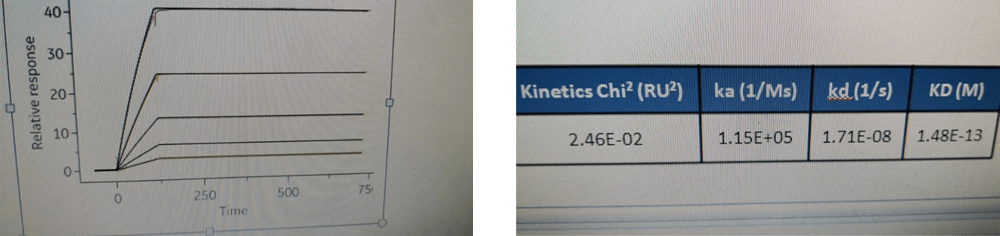
从图像上可以看出来没有饱和迹象,请降低捕获量,延长解离时间。
5. 筛选实验
用捕获法进行动力学的koff ranking(一般只看kd)的时候,我们capture已知浓度和分子量的抗体,然后走抗原分析物,这个时候除了kd比较准确外,同时看ka和KD准确吗?
抗体单浓度筛选时,一般都是基于kd进行排序。但仪器也会生成KD与ka,在kd接近时,也具有参考价值,也可以从KD的角度去做区分并进行Ranking。
6. 单循环动力学
在做单循环动力学的时候,最后一个浓度出现了曲线波动,请问这是什么原因造成的?

- 请检查以下事项最后一个样品的加样量够不够,
- 这个高浓度的样品有没有发生沉淀。